ATTR-kardiomyopatian syynä mutaatio luultua useammin
Lähtökohdat Kiinnostus transtyretiiniamyloidoosi (ATTR) -kardiomyopatiaa kohtaan on lisääntynyt parantuneen diagnostiikan ja taudinkulkua hidastavan lääkehoidon vuoksi.
Menetelmät Tutkimme pohjoismaisessa tutkimuksessa mukana olleen suomalaisen potilaskohortin (n = 19) oirekuvaa ja erityispiirteitä. Lisäksi arvioimme diagnoosien määrää kansallisissa hoitorekistereissä vuosina 2017–2021 sekä kahden TTR-geenimutaation esiintyvyyttä FinnGen-aineistossa.
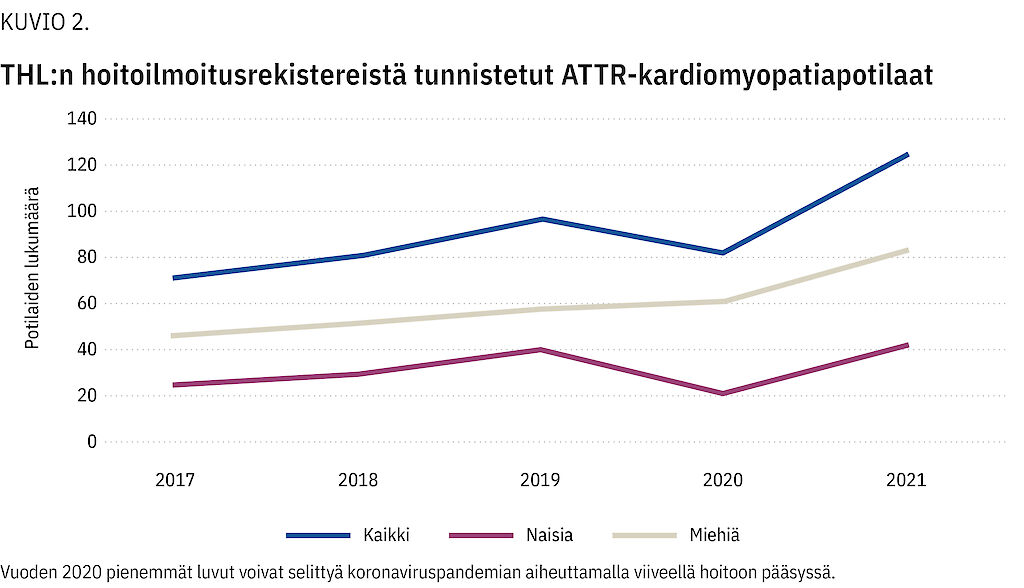
Tulokset Potilaiden keski-ikä oli 74,2 vuotta. Yleisimmät taudin ilmenemät olivat eteisvärinä (n = 13), sydämen vajaatoiminta (n = 10), rannekanavaoireyhtymä (n = 7) ja liitännäissairaus hypertensio (n = 7). Diagnoosien määrä kasvoi 1,7-kertaiseksi vuosina 2017–2021 ja ylitti 120 vuotuisen diagnoosin rajan vuonna 2021. Pelkästään TTR-mutaatioiden Val122Ile:n ja Val30Met:n esiintyvyys vastasi perinnöllisen taudin esiintyvyyttä eurooppalaisessa väestössä.
Päätelmät Suomalaispotilaiden oirekuva vastasi havaintoja muissa väestöissä. Diagnoosien määrän nopea kasvu viittaa sairauden alidiagnosointiin. Raportoimiemme kahden TTR-mutaation kantajien määrät Suomessa olivat oletettua suurempia. Tämä korostaa geenitestin tärkeyttä diagnostiikassa.

Transtyretiiniamyloidoosi (ATTR) on systeemisairaus, jossa elimistöön kertyy virheellisesti laskostunutta transtyretiinia (TTR). Sairauden tyypillisiä kliinisiä manifestaatioita ovat ATTR-kardiomyopatia ja perifeerinen neuropatia (1,2,3). Siitä tunnetaan perinnöllinen (ATTRm) ja hankinnainen (ATTRwt) muoto (2,3,4). Perinnöllisten TTR-geenimutaatioiden on uskottu olevan harvinaisia suomalaisessa väestössä (5).
Amyloidin kertyminen sydämeen aiheuttaa restriktiivisen kardiomyopatian. Tavallisia ovat voimakkaat oikean puolen vajaatoiminnan oireet ja löydökset, kuten hengenahdistus, alaraajaturvotus ja askites (1,6,7,8). Eteis-kammiojohtumisen häiriöitä esiintyy usein (8,9,10). Myös eteisvärinä on yleinen (1,6).
Muualla kehossa amyloidin kertyminen voi aiheuttaa perifeeristä tai autonomista neuropatiaa. Rannekanavaoireyhtymä on tyypillinen varhainen oire (1,2,6).
Elinajanodote oireiden alusta on 2–7 vuotta (7,8). Pohjoismaisen tutkimuksen mukaan potilaiden elämänlaatu on muita vajaatoimintapotilaita heikompi (11). Varhainen diagnoosi ja sairauden etenemistä hidastava lääkitys parantavat ennustetta ja elämänlaatua (9,10).
Kiinnostus ATTR-kardiomyopatiaa kohtaan on viime vuosina lisääntynyt, kun diagnostiikka ja lääkehoidot ovat kehittyneet. Gammakuvaus mahdollistaa amyloidoosin diagnoosin ilman koepalaa (12). Amyloidin kertymistä estävät hoidot ovat muuttaneet taudin luonnetta kroonisempaan suuntaan (13).
Tällä hetkellä taudinkulkuun vaikuttavista lääkkeistä Suomessa on käytössä TTR-proteiinin tetrameerirakennetta stabiloiva tafamidiisi. Tulevaisuudessa hoitoja saattavat olla myös RNA-interferenssiin tai antisense-oligonukleotideihin perustuvat TTR-proteiinin muodostumista estävät lääkkeet (14).
Suomalaisista ATTR-kardiomyopatiapotilaista on toistaiseksi vain vähän tietoa. Tässä tutkimuksessa selvitimme taudin oirekuvaa suomalaisilla potilailla, diagnoosimääriä vuosina 2017–2021 sekä kahden yleisimmän TTR-geenimutaation kantajafrekvenssiä suomalaisessa väestössä.
Potilaat ja menetelmät
Kliinisesti karakterisoidun tutkimusjoukon muodostivat 19 suomalaista ATTR-kardiomyopatiapotilasta. Heidät rekrytoitiin pohjoismaiseen Proact-tutkimukseen (n = 169) vuoden 2021 aikana kolmessa sairaalassa (Hus, Kys ja PKKS) (11). Sisäänottokriteereinä olivat yli 18 vuoden ikä ja vähintään kolme kuukautta kestänyt oireinen ATTR-kardiomyopatia. Jokainen potilas antoi kirjallisen suostumuksen tutkimukseen. Potilailta kerättiin demografiatiedot, oirekuva ja liitännäissairaudet.
Diagnoosit haettiin THL:n erikoissairaanhoidon ja perusterveydenhuollon hoitoilmoitusrekistereistä ajalta 2017–2021. Koska ATTR-kardiomyopatialla ei ole diagnoosikoodia Suomessa, haun kriteerinä olivat ennalta määritellyt ICD-10-diagnoosien yhdistelmät (E85.9*I43.1, E85*I42 tai E85*I50.9). Poissulkukriteerinä olivat diagnoosikoodit E85.80, C83.00, C88.0, C90 ja C91.1.
Kahden yleisimmän TTR-pistemutaation (Val122Ile ja Val30Met) kantajafrekvenssit selvitettiin suomalaisessa väestössä. Selvitys perustui FinnGen-tutkimusprojektin single nucleotide polymorphism (SNP) -genotyyppiaineiston (Release 12, yli 500 000 suomalaisen SNP-genotyyppitiedot) geenimerkkeihin rs76992529 ja rs28933979 (15).
Tulokset
Potilasaineiston ATTR-kardiomyopatiapotilaiden ikä oli keskimäärin 74,2 vuotta. Valtaosa (16/19) oli miehiä (taulukko 1).

Vain yhdellä potilaalla oli osoitettu TTR-geenin mutaatio (geenitieto puuttui seitsemältä). Eteisvärinä, sydämen vajaatoiminta ja rannekanavaoireyhtymä olivat taudin ilmenemistä yleisimpiä. Tyypillisin liitännäissairaus oli hypertensio.
Tiedonkeruuhetkellä diagnoosista oli keskimäärin 2,6 vuotta. Diagnostinen viive ensimmäisestä kardiologin tutkimuksesta diagnostiseen kuvantamiseen tai koepalaan oli keskimäärin 1,4 vuotta. Suurimmalla osalla sairaus oli diagnosoitu alkuvaiheessa (NYHA I-II). Ensimmäisinä oireinaan potilaat raportoivat yleisimmin hengenahdistuksen, fyysisen heikkouden tai huimauksen.
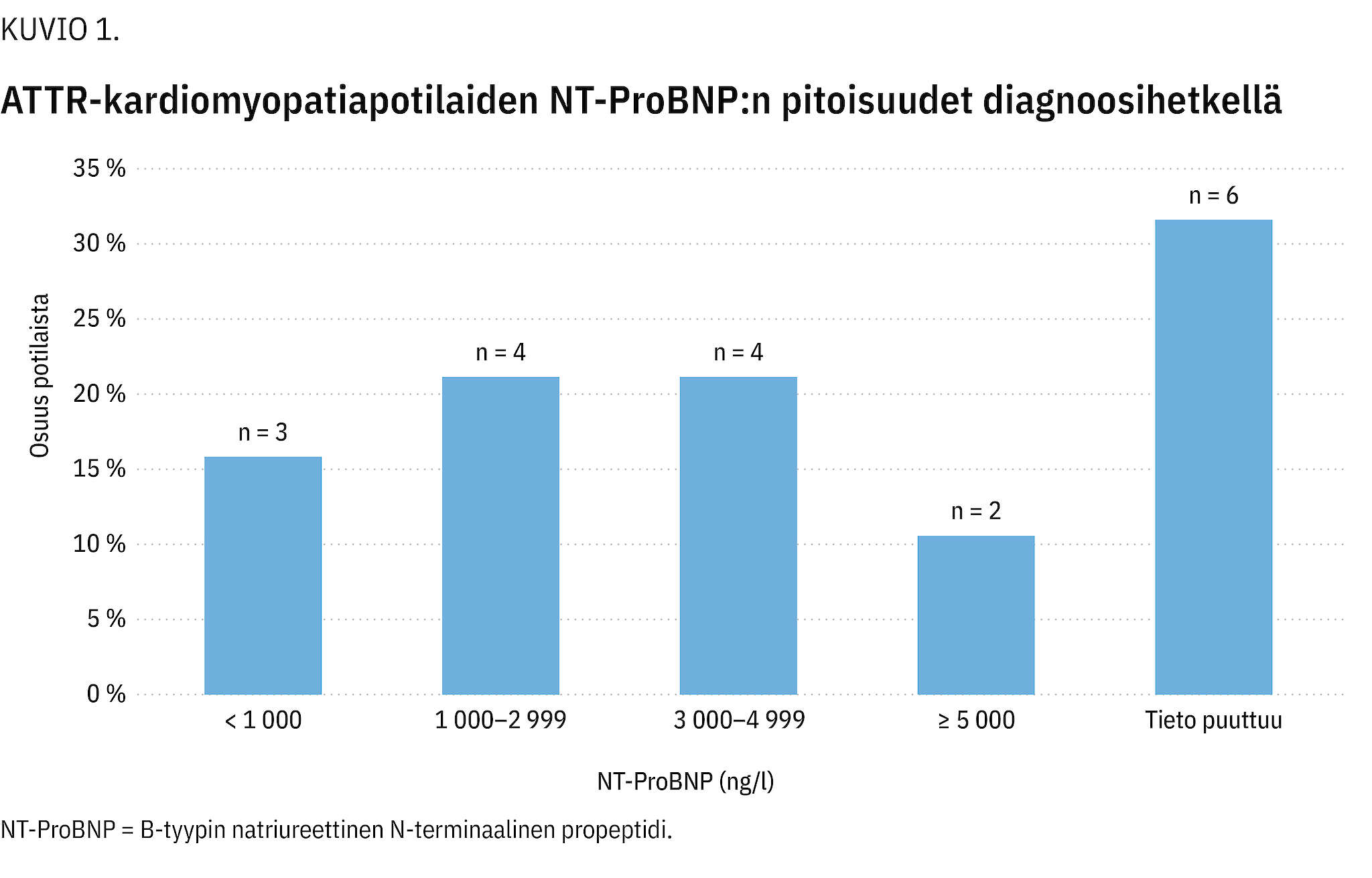
Osalta potilaista oli mitattu B-tyypin natriureettisen N-terminaalisen propeptidin pitoisuus (NT-ProBNP). Lähes puolella heistä arvo oli diagnoosihetkellä > 3 000 ng/l (kuvio 1). Vasemman kammion supistusvireys ja munuaisten toiminta olivat valtaosalla normaalit (taulukko 1).
Rekisterihaun perusteella diagnoosien määrä Suomessa kasvoi vuosina 2017–2021 (kuvio 2). Vuonna 2017 esiintyvyys oli 1,3 potilasta 100 000 henkilöä kohden. Vuonna 2021 luku oli jo 2,2. Naisia oli 34 %.
Kahden yleisimmän TTR-geenimutaation heterotsygootteja kantajia löytyi suomalaisesta FinnGen-aineistosta 20 (Val122Ile) ja 24 (Val30Met). Se vastaa väestössä kantajafrekvenssejä 2,01/100 000 ja 2,85/100 000.
Pohdinta
Suomalainen ATTR-kardiomyopatia vastaa taudinkuvia kansainvälisissä sarjoissa. Diagnoosimäärien kasvu Suomessa viime vuosina (2017–2021) on linjassa globaalin suuntauksen kanssa, jonka mukaan tauti tunnistetaan aiempaa paremmin.
Kansallisten hoitorekisterien tietojen mukaan naisten osuus diagnooseista oli 34 %. Luku eroaa eurooppalaisessa meta-analyysissa havaitusta osuudesta (17 %) (16).
Tutkimuskohorttiin rekrytoitiin vuoden aikana 19 suomalaispotilasta kolmessa sairaalassa. Määrä kuvaa ATTR-kardiomyopatiapotilaiden määrää erikoisairaanhoidossa.
Erityisesti varhaista diagnosointia tulisi edesauttaa ja kehittää, sillä uusista lääkehoidoista eivät hyödy pitkälle edennyttä tautia sairastavat, monisairaat tai hyvin iäkkäät potilaat.
FinnGen-aineiston perusteella TTR-mutaatiot ovat verrattain yleisiä suomalaisessa väestössä. Tämä haastaa aiemmat käsitykset niiden harvinaisuudesta (5). Osoitettujen mutaatioiden esiintyvyys tarkoittaa useampaa sataa mutaation kantajaa väestössä. Havainto tukee Euroopan kardiologisen seuran suositusta TTR-geenitestauksesta ATTR-kardiomyopatiadiagnoosin yhteydessä (8). Suosituksen mukaan perinnöllistä tautia sairastavien sukulaiset tulisi myös testata alaikäisiä lukuun ottamatta.
Tutkimus on ensimmäinen kuvaus suomalaisista ATTR-kardiomyopatiapotilaista sekä taudin esiintyvyydestä ja perinnöllisyydestä Suomessa. Hoitorekisterihaun rajoitteena on, että yksiselitteinen diagnoosikoodi puuttuu. Vain kahden yleisimmän TTR-mutaation esiintyvyys Suomessa oli mahdollista selvittää.
Lopuksi
Varttuneemman potilaan etenevän sydämen vajaatoiminnan taustalla voi olla ATTR-kardiomyopatia. Vaikka tautia on pidetty pääasiallisesti iäkkäämpien miesten tautina, sen mahdollisuus on tärkeää huomioida myös naisilla.
Diagnoosimäärien nopea kasvu viittaa sairauden alidiagnosointiin. Säännöllinen geenitestaus auttaa löytämään oireettomia ja vähäoireisia sukulaisia, sillä TTR-mutaation kantajia on enemmän kuin aiemmin on ajateltu.
Kiitämme Proact-tutkimukseen osallistuneita potilaita, sydämen vajaatoimintahoitaja Julia Raak-Tarkiaista (Hus Sydän- ja keuhkokeskus) avusta Proact-tutkimuksen tiedonkeruussa ja Sanna Hellbergiä (MedEngine oy) avusta käsikirjoituksen työstämisessä.
Nordic Proact-tutkimuksen on sponsoroinut Pfizer. Mira Kyttälä on Pfizer oy:n palveluksessa ja omistaa yhtiön osakkeita. Aino Vesikansa on MedEngine oy:n työntekijä ja saanut rahoitusta Pfizer oy:ltä tämän artikkelin kirjoittamiseen. Kilpailevia eturistiriitoja ei ole ilmoitettavana.
Kirjoittajien ilmoittama käsikirjoitukseen liittyvä rahoitus: Pfizer oy.
Riina Kandolin: Konsultointi (Pfizer), luentopalkkiot (Pfizer).
Johanna Kuusisto: Konsultointi (Pfizer), apuraha (Pfizer), luentopalkkiot (Pfizer).
Kati Valtola: Apuraha (Pfizer).
Aino Vesikansa: Korvaus käsikirjoituksen kirjoittamisesta (Pfizer).
Jukka Lehtonen: Luentopalkkiot (Pfizer).
Mira Kyttälä, Tuomas Rissanen: Ei sidonnaisuuksia.
- 1
- Rapezzi C, Quarta CC, Riva L ym. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol 2010;7:398–408.
- 2
- Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012;126:1286–300.
- 3
- González-López E, López-Sainz Á, Garcia-Pavia P. Diagnosis and treatment of transthyretin cardiac amyloidosis. Progress and hope. Rev Esp Cardiol Engl Ed 2017;70:991–1004.
- 4
- Maurer MS, Hanna M, Grogan M ym. Genotype and phenotype of transthyretin cardiac amyloidosis. J Am Coll Cardiol 2016;68:161–72.
- 5
- Pettersson T. Edistystä systeemisen amyloidoosin diagnostiikassa ja hoidossa. Suom Lääkäril 2017;72:1353–8.
- 6
- Holkeri A, Pettersson T, Mustonen T, Holmström M, Atula S, Lehtonen J. Sydänamyloidoosi. Duodecim 2019;135:2291–9.
- 7
- Gertz MA, Benson MD, Dyck PJ ym. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol 2015;66:2451–66.
- 8
- Garcia-Pavia P, Rapezzi C, Adler Y ym. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2021;42:1554–68.
- 9
- Rozenbaum MH, Large S, Bhambri R ym. Estimating the health benefits of timely diagnosis and treatment of transthyretin amyloid cardiomyopathy. J Comp Eff Res 2021;10:927–38.
- 10
- Maurer MS, Schwartz JH, Gundapaneni B ym. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007–16.
- 11
- Eldhagen P, Lehtonen J, Gude E ym. Health‐related quality of life among transthyretin amyloid cardiomyopathy patients. ESC Heart Fail 2023;ehf2.14350.
- 12
- Gillmore JD, Maurer MS, Falk RH ym. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016;133:2404–12.
- 13
- Tschöpe C, Elsanhoury A. Treatment of transthyretin amyloid cardiomyopathy: the current options, the future, and the challenges. J Clin Med 2022;11:2148.
- 14
- Aimo A, Castiglione V, Rapezzi C ym. RNA-targeting and gene editing therapies for transthyretin amyloidosis. Nat Rev Cardiol 2022;19:655–67.
- 15
- Kurki MI, Karjalainen J, Palta P ym. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 2023;613:508–18.
- 16
- Bruno M, Castaño A, Burton A, Grodin JL. Transthyretin amyloid cardiomyopathy in women: frequency, characteristics, and diagnostic challenges. Heart Fail Rev 2021;26:35–45.
- 17
- Dolgin M, the Criteria Committee of the New York Heart Association, Fox AC, Gorlin R, Levin RI. Nomenclature and criteria for diagnosis of diseases of the heart and great vessels. 9. painos. Boston, MA: Lippincott Williams and Wilkins 1994.
- 18
- Gillmore JD, Damy T, Fontana M ym. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018;39:2799–806.
Mutation as the cause of ATTR cardiomyopathy is more common than believed
Background Interest in transthyretin amyloid cardiomyopathy (ATTR-CM) is growing due to improved diagnostics and disease modifying therapy.
Methods We investigated the symptoms and disease characteristics in a Finnish ATTR-CM patient cohort participating in a Nordic study (n=19). We also evaluated the development of ATTR-CM diagnosis numbers in national care registries in 2017–2021 and the prevalence of two known TTR gene mutations using the FinnGen register.
Results The mean age of patients was 74.2 years. The most common disease manifestations were atrial fibrillation (n=13), heart failure (n=10), and carpal tunnel syndrome (n=7), and the most common comorbidity was hypertension (n=7). The number of ATTR-CM diagnoses increased by 1.7-fold during the years 2017–2021, exceeding 120 annual diagnoses in 2021. In the population data, the prevalence of TTR mutations Val122Ile and Val30Met corresponded to the prevalence of hereditary ATTR-CM in European population.
Conclusions The symptoms of Finnish ATTR-CM patients corresponded to the observations in other populations. The rapid increase in diagnoses is indicative of underdiagnostics. The numbers of carriers of the two examined TTR mutations in Finland were higher than expected, highlighting the importance of genetic testing in diagnostics.