Primaarinen immuunivajavuus: harvinaisia sairauksia, joiden tunnistaminen on tärkeää
• Primaarinen immuunivajavuus kattaa joukon synnynnäisiä immuunipuolustuksen häiriöitä, joiden oirekirjo on laaja ja vaihteleva jopa saman geenimutaation kantajilla.
• Oireiden ilmaantumisikä vaihtelee imeväisestä aikuisikään.
• Primaarinen immuunivajavuus on mahdollinen, jos potilaalla on toistuvia tai epätyypillisiä infektioita, varsinkin jos näihin liittyy autoimmuuni- tai autoinflammatorisia oireita.
• Suvussa esiintyvä immuunivajavuus tai suvun poikkeava infektiohistoria voi viitata primaariseen immuunivajavuuteen.
• Vakavissa lapsuuden immuunivajeissa kantasolusiirto on usein hengen pelastava toimenpide.

Primaarinen immuunivajavuus käsittää joukon harvinaisia sairauksia, jotka aiheuttavat immuunipuolustuksen merkittävän häiriön. Kansainvälinen immunologian asiantuntijajärjestö (The International Union of Immunological Societies, IUIS) julkaisee joka toinen vuosi näistä raportin. Vuoden 2013 raportissa oli 250 geenivirhettä, jotka aiheuttivat monogeenisen immuunivajavuuden, vuonna 2019 niitä oli jo 430 (1).
Eurooppalaisessa primaaristen immuunivajavuuksien rekisterissä oli 29 527 potilasta vuonna 2021 (taulukko 1) (2).

Prevalenssi sataatuhatta asukasta kohti on rekisteritietojen perusteella Saksassa 2,7, Ranskassa 11,0 ja Britanniassa 5,9 (3,4,5). Prevalenssi on kasvanut viime vuosikymmeninä. Merkittävä syy on potilaiden parempi tunnistaminen, kun tietoisuus primaarisista immuunivajavuuksista on lisääntynyt.
Suomessa ei ole kansallista rekisteriä, mutta suomalaiseen perimään on rikastunut useita immuunivajavuutta aiheuttavia geenivariantteja, esimerkiksi AIRE (APECED) ja rusto-hiushypoplasia. Lisäksi aikuisen yleisen vaihtelevan immuunivajavuuden (common variable immunodeficiency, CVI) esiintyvyys on suuri, joten primaaristen immuunivajavuuksien prevalenssi on todennäköisesti vähintään yhtä suuri kuin Euroopassa (6,7). Tätä tukee aikuisten infektiopoliklinikan seurannassa olevien potilaiden perusteella arvioitu prevalenssi Pirkanmaalla, noin 14/100 000 asukasta.
Milloin epäillä primaarista immuunivajavuutta?
Toistuvat infektiot kuuluvat lapsuuteen, ja aikuisellakin niitä esiintyy muutamia vuosittain, vaikka immuniteetti on normaali. Päivähoidossa oleva lapsi voi sairastaa jopa 10 hengitystieinfektiota vuodessa, ja lähes jokainen suomalainen lapsi saa antibioottihoidon hengitystieinfektioon kolmeen ikävuoteen mennessä (8). Tämä infektiopaine näkyy usein lähipiirin aikuisten lisääntyneinä ylähengitystieinfektioina.
Infektioita lisäävät infektiopaineen lisäksi mm. ikääntyminen, elintavat (tupakointi ja runsas alkoholin käyttö), aliravitsemus, akuutit ja pitkäaikaissairaudet sekä immuniteettiin vaikuttavat hoidot. Sisätautiosastojen potilaista vähintään kolmella neljästä on jokin immuniteettia heikentävä tekijä.
Jos infektioista paranee hyvin ilman suonensisäistä antibioottihoitoa eikä sairasta syviä infektioita ja lapsella kasvu jatkuu normaalisti, on immunologinen puolustus todennäköisesti kunnossa.
Immuunivajavuuspotilaita hoitavat lääkärit ovat julkaisseet asiantuntijamielipiteeseen perustuvia varoitusmerkkejä, joiden avulla on pyritty primaarista immuunivajavuutta sairastavien yksilöiden tunnistamiseen (taulukko 2).

Varoitusmerkit ovat lisänneet tietoisuutta, mutta primaariset immuunivajavuudet ovat niin harvinaisia ja taudinkuva monimuotoinen, että niiden tunnistaminen on haastavaa (9,10,11,12).
Varoitusmerkkejä tutkittaessa toimiviksi ovat osoittautuneet huono painonkehitys, lymfopenia, pitkittynyt suonensisäisen antibiootin tarve ja suvussa esiintyvä immuunipuutos (7,9). Saksan ja Britannian immuunivajavuusrekisterin potilaista peräti 21 %:lla ja 24 %:lla oli suvussa immuunivajavuusr ja 8 %:lla ja 2,9 %:lla vanhemmat olivat sukulaisia (3,5).
Lisävaroitusmerkkejä on kehitetty kliinisten erikoisalojen lääkäreille avuksi diagnostiikassa (13,14,15). Niiden perusteella on keskeistä epäillä primaarista immuunivajavuutta, jos taudinkuva on monimuotoinen, siihen liittyy autoimmuuni- tai autoinflammatorisen taudinkuvan piirteitä, varsinkin jos ne yhdistyvät infektioihin.
Oireet ja taudinkuva auttavat diagnoosin jäljille
Thalhammer ym. analysoivat Euroopan rekisteriin hyväksyttyjen potilaiden diagnoosiin johtaneet ensioireet (16). Infektio oli ensioireena 77 %:lla 16 486 potilaasta, immunodysregulaatio 18 %:lla, sopiva taudinkuva 12 %:lla ja maligniteetti 0,8 %:lla. Immunodysregulaation taudinkuva piti sisällään mm. lymfoproliferaation, inflammatoriset suolisto-oireet, vaskuliitin, ekseeman, kuumeen ja autoimmuunioireet. Joka viidennellä potilaalla oli useita ensioireita; tavallisin yhdistelmä oli infektio ja immunodysregulaatio. Huomionarvoista on, että 23 %:lla ensioire ei ollut infektio ja yli 10 %:lla potilaista ei ollut lainkaan infektio-oireita. Diagnoosiin johtavat oireet alkoivat 76 %:lla lapsuusiässä, 33 %:lla alle 1 vuoden ja 30 %:lla 1–5 vuoden iässä. Yli 40-vuotiaana oireet alkoivat vain 9 %:lla.
Primaarista immuunivajavuutta sairastavia potilaita on niin perusterveydenhuollon kuin eri erikoisalojen ongelmapotilaissa. Pirkanmaan sairaanhoitopiirissä diagnosoiduista aikuispotilaista viidesosa on siirtynyt seurantaan lastenklinikasta ja neljäsosa avohoidosta, mutta suurin osa eri erikoisalojen konsultaationa. Oireet vaihtelevat henkeä uhkaavista lievää terveyshaittaa aiheuttaviin. Vaikka geenimutaatio olisi sama, taudin ilmiasu ja vaikeusaste voivat olla erilaiset. Vaikeimmat tautimuodot ilmenevät yleensä lapsuusiässä, mutta ne voivat myös ilmaantua vasta aikuisiällä immuunijärjestelmän ”vanhentuessa”.
Vaikka primaarisia immuunivajavuuksia löytyy yhä useammin potilailta, joilla on autoimmuunitauti tai immuunisäätelyn häiriö, infektio on tärkein diagnoosiin johtava oire (taulukko 3) (16). Ikä oireiden alkaessa auttaa diagnostiikassa.
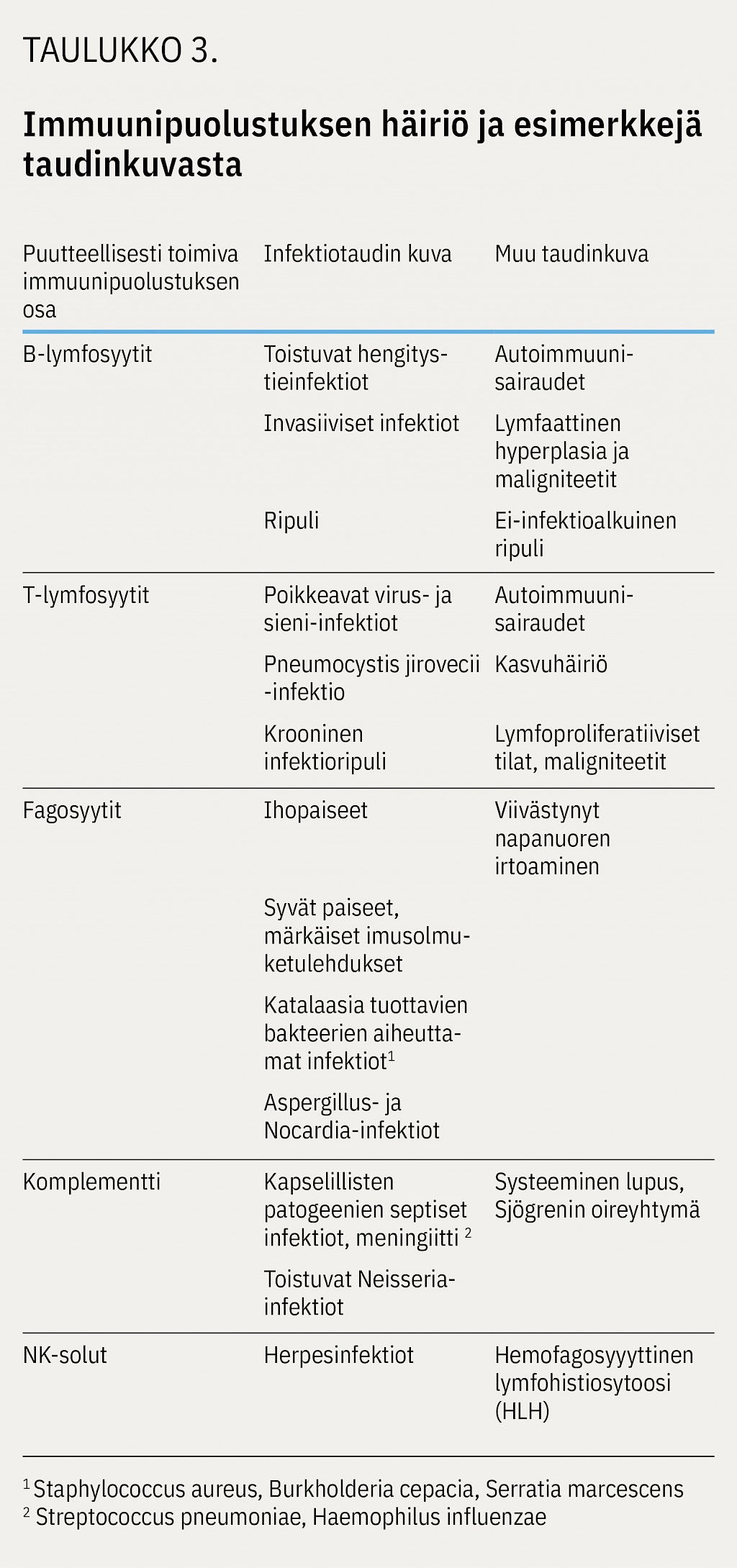
Vakavimmat immuunipuutokset, kuten vaikea sekamuotoinen immuunivajavuus eli SCID-oireyhtymä, synnynnäinen neutropenia, hemofagosyyttinen lymfohistiosytoosi ja osa autoinflammatorisista ja immunodysregulaation oirekuvista, oireilevat ensimmäisen ikävuoden aikana. Vaikeat vasta-ainepuutokset alkavat oireilla toistuvina hengitystie- tai septisinä infektioina 3–9 kuukauden iässä, kun lapsen äidiltä istukan kautta saamat vasta-aineet häviävät eikä oma tuotanto käynnisty (17). Neutrofiilien toimintahäiriö, krooninen granulomatoosi, diagnosoidaan usein leikki-iässä samoin kuin vakavien immuunivajavuuksien lievemmät oirekuvat.
Suuri osa diagnoosiin johtavista infektioista on tavallisten patogeenien aiheuttamia toistuvia tai yksittäisiä poikkeavia infektioita (18). Joskus immuunivajavuuden aiheuttava geenivirhe on niin spesifinen, että se altistaa vain yhden patogeenin aiheuttamille infektioille (taulukko 4) (1,18,22).
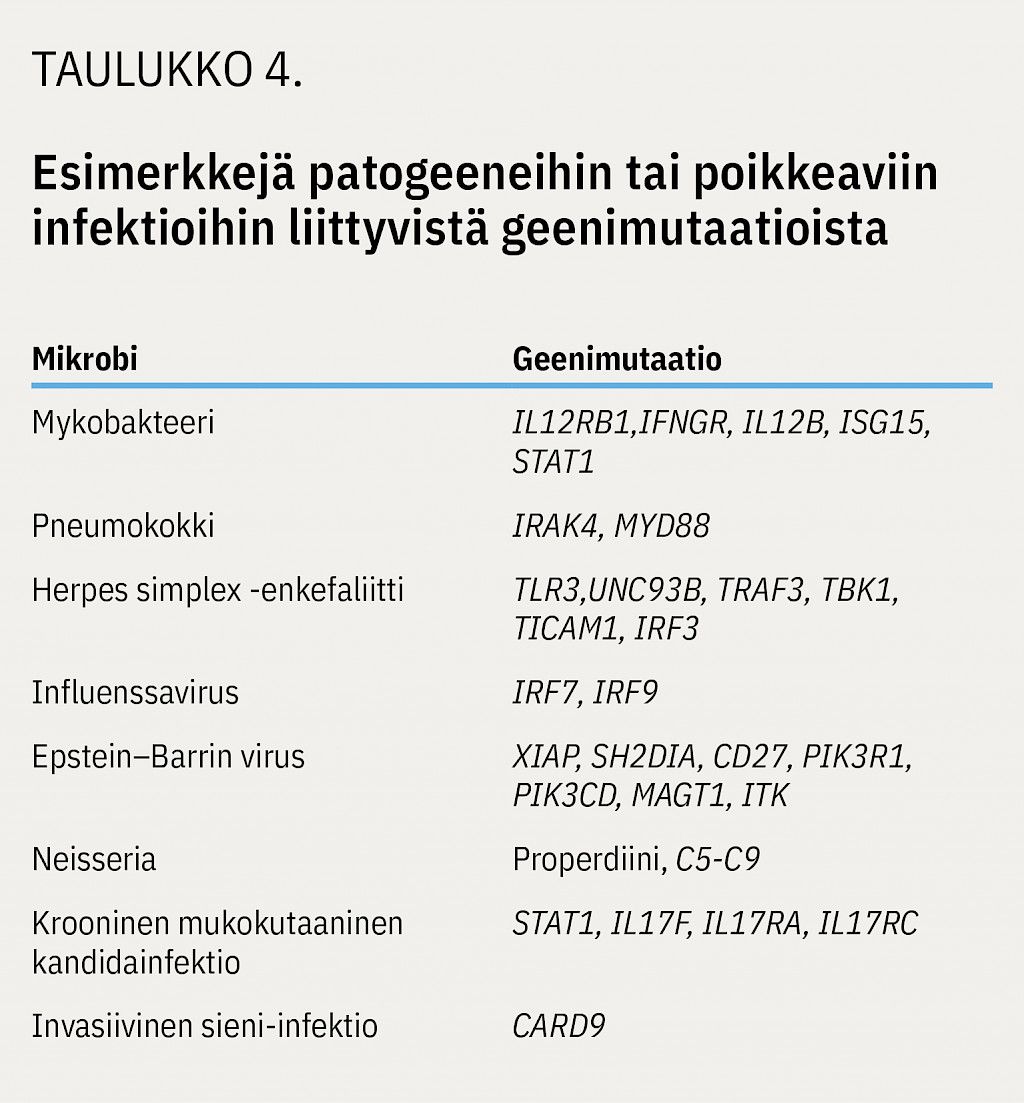
Vanhemmalla lapsella toistuvien infektioiden takaa voi löytyä yleinen vaihteleva immuunivajavuus eli CVI-tauti, jossa B-soluja löytyy mutta niiden toiminta ei ole optimaalista (1). Tähän tautiryhmään kuuluu suurin osa aikuisiällä diagnosoiduista potilaista (16). Pirkanmaan sairaanhoitopiirin aikuisten infektiopoliklinikan potilaista 80 % on saanut diagnoosin yli 16-vuotiaana, vanhin yli 80-vuotiaana.
Immuunivajavuuden tunnistaminen on tärkeää
Varhain aloitettu hoito on keskeistä potilaan ennusteen kannalta. Parhaiten tämä on osoitettu vaikeassa synnynnäisessä immuunivajavuudessa: alle 3 kuukauden iässä tehty kantasolusiirto vähentää kuolleisuutta verrattuna tehdään yli 6 kuukauden iässä tehtyyn siirtoon, jolloin lapsi on jo sairastunut ensimmäiseen infektioon ja komplikaatioriski kantasolusiirrossa on merkittävä (19).
Imeväiset, joilla on vakava T- ja B-solujen toimintahäiriö, vaikea synnynnäinen immuunivajavuus, sairastavat pitkittyneitä infektioita, heillä voi olla hankala ihottuma ja huono menestyminen. Oireet voi alkuvaiheessa olla vaikea erottaa suolisto- ja iho-oireisesta allergiasta.
Turussa vuonna 2019 käynnistyneen ja viime vuonna koko maahan laajentuneen vastasyntyneiden immuunipuutosseulonnan tavoitteena on löytää vaikeat synnynnäiset immuunivajavuudet ennen ensimmäistä infektiota. Seulonnassa mitataan 2–5 vrk:n iässä vauvasta otetusta veritäplänäytteestä T-solureseptoreja muodostettaessa syntyvien DNA-kopioiden (T-cell receptor excision circles, TREC) määrää. Vaikeassa synnynnäisessä immuunivajavuudessa TREC-tasot ovat matalat. TREC-mittaukseen perustuvaa seulontaa on käytetty Yhdysvalloissa vuodesta 2008 alkaen, ja se löytää sairaat vastasyntyneet hyvin (20).
Myös myöhemmin lapsuudessa ja aikuisiässä saatu diagnoosi parantaa ennustetta. Suomessa yleisen vaihtelevan immuunivajavuuden, CVI-taudin, esiintyvyys on suuri, ja näiden potilaiden löytäminen ja heille räätälöity immunoglobuliinikorvaushoito pidentää eliniän ennustetta kymmenillä vuosilla (7,21,22).
Immuunivajavuusepäilyn arvioimisessa anamneesin merkitys korostuu. Vanhemmilla lapsilla ja aikuisilla immunoglobuliinien määrittäminen on halpa menetelmä vasta-ainepuutosten seulontaan. Jos oireet ja löydökset vaikuttavat poikkeuksellisilta, vaikka perusterveydenhuollossa katsotut tutkimukset olisivat normaaleja, eikä taustalta löydy selittäviä tekijöitä, tulee potilaasta konsultoida erikoissairaanhoitoa. Primaaristen immuunivajavuuksien diagnostiikka, tutkimukset ja hoidon suunnittelu on keskitetty erikoissairaanhoitoon. Vaikka immuunivajavuuteen ei olisi täsmähoitoa tarjolla, sairastavuuteen voidaan vaikuttaa optimoimalla liitännäissairauksien hoitoa sekä muita tekijöitä, kuten ravitsemus, rokotteet (myös lähipiirin), suun ja ihon terveys. Diagnoosin asettamisella vältetään myös toistuva tutkimuskierre.
Geenidiagnostiikan tuomat mahdollisuudet
Kansainvälinen luokitus jakaa immuunivajavuudet 10 luokkaan kliinisen taudinkuvan ja potilaan immunologisen löydöksen perusteella (23). Immunologisten tulosten tulkinta ei ole aina suoraviivaista, ja yhteistyö laboratorion ja kliinisten asiantuntijoiden kanssa on tärkeää. Esimerkiksi pienillä lapsilla lymfosyyttitasot ovat korkeammat kuin aikuisilla, joten laboratorion tuottamiin aikuisviitearvoihin luottaminen voi johtaa diagnoosin viivästymiseen (24).
Geeni- ja solutason tutkimusmenetelmien kehittyminen on mahdollistanut taustalla olevien geneettisten syiden ja immunologisten mekanismien tarkentamisen. Poikkeavien taudinkuvien takaa löytyy uusia geenimutaatioita, ja aikaisemmin tunnettuihin mutaatioihin liitetyt taudinkuvat monipuolistuvat.
Geenidiagnostiikkaa voidaan tehdä sekvensoimalla kohdennetusti kandidaattigeeni, jos diagnostinen epäily on vahva tai suvun geenivirhe on tiedossa. Jos diagnostisia vaihtoehtoja on useita, voidaan käyttää kaupallisia geenipaneeleja, jotka sisältävät jopa satoja tunnettuja geenejä. Jos aiheuttajageeni on tuntematon ja epäily immuunivajavuudesta on vahva tehdään eksomisekvensointi (25,26). Koko genomin sekvensointi on vielä lähinnä tutkimuskäytössä.
Kansainvälinen yhteistyö on tärkeää, kun geenipoikkeavuus on uusi tai se on aikaisemmin kuvattu vain yksittäisessä perheessä (1). Taudin aiheuttajageenin ja sen mutaation tunnistaminen mahdollistaa sikiödiagnostiikan, taudin ennusteen arvioinnin ja vähentää kliinikon ja perheen epävarmuutta ja turhia tutkimuksia (27).
Immuunivajavuutta selittävää geenipoikkeavuutta ei kuitenkaan löydy kuin osalta potilaista. Vakavissa lapsuusiässä oireilevissa primaarisissa immuunivajavuuksissa geenipoikkeavuuden löytyminen on todennäköisempää kuin aikuisiällä.
Saksan immuunipuutosrekisterissä 36 %:lla kaikista ja 84 %:lla geenitestatuista löytyi oirekuvan selittävä geenimutaatio (3). Vaikeaa synnynnäistä immuunivajavuutta sairastavista imeväisistä 75 %:lta löytyi geenimutaatio. CVI-potilaista 70 %:a ei ollut testattu, mutta niistä, joille geenidiagnostiikkaa oli tehty, 30 %:lta löytyi oirekuvan selittävä mutaatio (3). Euroopan rekisterin potilaista 26 %:lla (n = 4 228) oli CVI ja heistä vain 0,03 %:lla oli geenidiagnoosi (16).
Lopuksi
Primaarista immuunivajavuutta sairastavien potilaiden tunnistaminen on haastavaa mutta tärkeää. Potilaiden oireet ovat kroonisia ja voivat hoitamattomina johtaa elämänlaadun heikkenemiseen ja ennenaikaiseen kuolemaan.
Geenidiagnostiikan kehittyminen, parantunut saatavuus ja tutkimusten hintojen lasku ovat tehostaneet immuunivajavuuspotilaiden diagnostiikkaa ja lisänneet uusien tautien ja niitä aiheuttavien geenien tunnistamista. Vuonna 2016 Euroopan lääkevirasto hyväksyi EU-alueella geeniterapian vaikean synnynnäisen immuunivajavuuden alatyypin, adenosiinideaminaasipuutoksen, hoitoon (28).
Immuunivajavuuksia aiheuttavien tautien, niiden takana olevien geenivirheiden tunnistaminen ja solutason mekanismien tunnistaminen mahdollistaa uusien lääkkeiden kehittämisen. Näitä täsmälääkkeitä on yhä enemmän käytössä potilailla, jotka sairastavat eri immunologisilla mekanismeilla syntyviä sairauksia (29,30,31,32).
Kiitokset dosentti Jaana Syrjäselle käsikirjoituksen kommentoimisesta.
Merja Helminen, Hanna Viskari: Ei sidonnaisuuksia.
- 1
- Tangye SG, Al-Herz W, Bousfiha A ym. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2020;40:24–64.
- 2
- Data from the ESID online database (www.esid.org).
- 3
- El-Helou SG, Biegner A-K, Bode S ym. The German national registry of primary immunodeficiencies (2012-2017). Front Immunol 2019;10:1272.
- 4
- Mahlaoui N, Jais J-P, Brosselin P ym. Prevalence of primary immunodeficiencies in France is underestimated. J Allergy Clin Immunol 2017;140:1731–3.
- 5
- Shillitoe B, Bangs C, Guzman D ym. The United Kingdom primary immune deficiency (UKPID) registry 2012 to 2017. Clin Exp Immunol 2018;192:284–91.
- 6
- Grönholm J, Seppänen M. Immuunivajeiden mekanismit – uuden äärellä. Duodecim 2020;136:2591–600.
- 7
- Selenius JS, Martelius T, Pikkarainen S ym. Unexpectedly high prevalence of common variable immunodeficiency in Finland. Front Immunol 2017;8:1190.
- 8
- Toivonen L, Karppinen S, Schuez-Havupalo L ym. Burden of recurrent respiratory tract infections in children. Pediatr Infect Dis J 2016;35:e362–9.
- 9
- MacGinnitie A, Aloi F, Mishra S. Clinical characteristics of pediatric patients evaluated for primary immunodeficiency. Pediatr Allergy Immunol 2011;22:671–5.
- 10
- Subbarayan A, Colarusso G, Hughes SM ym. Clinical features that identify children with primary immunodeficiency diseases. Pediatrics 2011;127:810–6.
- 11
- Arslan S, Ucar R, Caliskaner AZ ym. How effective are the 6 European Society of Immunodeficiency warning signs for primary immunodeficiency disease? Ann Allergy Asthma Immunol 2016;116:151–5.
- 12
- Lankisch P, Schiffner J, Ghosh S ym. The Duesseldorf warning signs for primary immunodeficiency: is it time to change the rules? J Clin Immunol 2015;35:273–9.
- 13
- Carneiro-Sampaio M, Jacob CMA, Leone CR. A proposal of warning signs for primary immunodeficiencies in the first year of life. Pediatr Allergy Immunol 2011;22:345–6.
- 14
- O’Sullivan MD, Cant AJ. The 10 warning signs: a time for a change? Curr Opin Allergy Clin Immunol 2012;12:588–94.
- 15
- Costa-Carvalho B, Grumach AS, Franco JL ym. Attending to warning signs of primary immunodeficiency diseases across the range of clinical practice. J Clin Immunol 2014;34:10–22.
- 16
- Thalhammer J, Kindle G, Nieters A ym. Initial presenting manifestations in 16,486 patients with inborn errors of immunity include infections and noninfectious manifestations. J Allergy Clin Immunol 2021;148:1332–41.e5.
- 17
- Vetrie D, Vorechobsky I, Sideras P ym. The gene involved in X-linked agammaglobulinemia is a member of the src family of protein- tyrosine kinases. Nature 1993;361:226–33.
- 18
- Casanova J-L. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci USA 2015;112:E7128–37.
- 19
- Pai SY, Logan BR, Griffith LM ym. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med 2014;371:434–46.
- 20
- Dorsey M, Puck J. Newborn screening for severe combined immunodeficiency in the US: current status and approach to management. Int J Neonatal Screen 2017;3:15.
- 21
- Perez E, Orange J, Bonilla F ym. Update on the use of immunoglobulin in human disease: A review of evidence. J Allergy Clin Immunol 2017;139 (3S): S1–S46.
- 22
- Kainulainen L, Nikoskelainen J, Ruuskanen O. Diagnostic findings in 95 Finnish patients with common variable immunodeficiency. J Clin Immunol 2001;21:145–9.
- 23
- Bousfiha A, Jeddane L, Picard C ym. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol 2020;40:66–81.
- 24
- Van Gent R, van Tilburg CM, Nibbelke EE ym. Refined characterization and reference values of the pediatric T- and B-cell compartments. Clin Immunol 2009;133:95–107.
- 25
- Stray-Pedersen A, Sorte HS, Samarakoon P ym. Primary immunodeficiency diseases: Genomic approaches delineate heterogenous Mendelian disorders. J Allergy Clin Immunol 2017;139:232–45.
- 26
- Arts P, Simons A, AlZahrani MS ym. Exome sequencing in routine diagnostics: a generic test for 254 patients with primary immunodeficiencies. Genome Med 2019;11:38.
- 27
- Elsink K, van Montfrans JM, van Gijn ME ym. Cost and impact of early diagnosis in primary immunodeficiency disease: a literature review. Clin Immunol 2020;213:108359.
- 28
- Ferrari G, Thrasher AJ, Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nature 2021;22:216–34.
- 29
- Chellapandian D, Chitty-Lopez M, Leiding JW. Precision therapy for the treatment of primary immunodysregulatory diseases. Immunol Allergy Clin North Am 2020;40:511–26.
- 30
- De Benedetti F, Gattorno M, Anton J ym. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med 2018;378:1908–19.
- 31
- Damsky W, Peterson D, Ramseier J ym. The emerging role of Janus kinase inhibitors in the treatment of autoimmune and inflammatory diseases. J Allergy Clin Immunol 2021;147:814–26.
- 32
- Ruiz-Garcia R, Vargas-Hernandez A, Chinn IK ym. Mutations in PI3K110 λ cause impaired NK cell function partially rescued by rapamycin treatment. J Allergy Clin Immunol 2018;142:605–17.
Primary immunodeficiency diseases: rare disorders where early recognition is important
Primary immunodeficiencies (PIDs) or inborn errors of immunity (IEIs) are diseases with broad clinical manifestations. In 2013 the International Union of Immunological Societies Expert Committee on Inborn Errors of Immunity reported 250 monogenic diseases while in 2019 the number had increased to 430.
Warning signs, based on clinical presentation and expert opinion, have been published to increase awareness and early diagnosis of PIDs. In some studies only a family history of PID, poor growth, prolonged need for iv antibiotics and lymphopenia have been shown to find the PID patients. The most severe PIDs, such as severe combined immunodeficiency (SCID), present in early infancy. Early diagnosis and prompt stem cell transplantation is crucial for the babies’ survival. Newborn screening for SCID was implemented in Finland this year. Screening is based on quantification of T-cell receptor excision circles, which are absent or very low in SCID babies. The sample is obtained from the infant at 3–5 days of age and the dried blood spots analysed with PCR. Immune deficiencies diagnosed later in life include humoral immune deficiencies, neutrophil disorders, complement deficiencies and the adult common variable immunodeficiency (CVID). Increasingly, PIDs with atypical clinical pictures, immune dysregulation and autoimmune features are being diagnosed. The developments in molecular diagnostics and cell technology have shaken the field. New genes are being discovered, new phenotypes connected to known genes and immunological mechanisms detected. Novel treatments have been developed for PID patients as well as for patients with polygenic autoimmune and autoinflammatory diseases.
Merja Helminen, Hanna Viskari
Merja Helminen
Senior Lecturer, Deputy Chief Physician
Department of Paediatrics, Tampere University Hospital