Kuinka yleisiä ovat lihastaudit Suomessa?
• Lihastautauteja sairastavien tarkkaa määrää Suomessa ei tiedetä. Kirjallisuuden perusteella luvun arvioidaan voivan olla jopa lähes 17 000.
• Uudet, kalliit hoidot vaativat huolellista seurantaa.
• Kattavalla kansallisella lihastautirekisterillä olisi suuri merkitys hoidon kehittämisen, kansainvälisen yhteistyön ja resurssien suuntaamisen kannalta.

Lihastaudit eli neuromuskulaaritaudit ovat heterogeeninen ryhmä harvinaistauteja, joita sairastavien määrästä Suomessa ei ole tarkkoja tietoja. Ryhmään kuuluvat sekä myopatiat eli itse lihaskudoksen taudit että hermo-lihasliitoksen taudit, motoneuronitaudit ja perifeeriset, motorisia hermoja vaurioittavat polyneuropatiat.
Tämä katsaus perustuu uusimpiin epidemiologisiin lähteisiin, joiden perusteella on laskettu arviot lihastauteja sairastavien henkilöiden lukumääristä Suomessa. Tiedolla on huomattavaa käytännön merkitystä.
Lihastaudit ovat harvinaisia sairauksia, eli niitä sairastaa harvempi kuin 1:2 000. Osa on erittäin harvinaisia tauteja, joita sairastaa harvempi kuin 1:50 000. Suuri osa neuromuskulaaritaudeista on geenivirheen aiheuttamia, mutta osa on hankinnaisia.
Taudinkuvat vaihtelevat vähäoireisista vaikeisiin, eteneviin ja ennenaikaiseen kuolemaan johtaviin sairauksiin. Osa taudeista ilmenee lapsuudessa, joskus jopa vastasyntyneellä, osa nuoruudessa ja työiässä, mutta moni tauti tulee esiin vasta ikävuosien karttuessa ja sairastavuus painottuu vanhusväestöön.
Yhteistä näille taudeille on, ettei näihin päiviin asti ole ollut olemassa parantavaa hoitoa. Lastenneurologit ja aikuisneurologit ovat vastuussa lihastautien diagnostiikasta ja erikoissairaanhoidosta. Hoito on monessa vaikeassa taudissa, kuten Duchennen lihasdystrofiassa (DMD) tai motoneuronitaudeissa eli spinaalisessa lihasatrofiassa (SMA) ja amyotrofisessa lateraaliskleroosissa (ALS), haastavaa ja usean erikoisalan osaamista vaativaa tiimityötä. Sen painopisteenä on päivittäisen arjen helpottaminen kuntoutuksen avulla. Apuvälineillä, joihin kuuluvat mm. rollaattori, pyörätuoli, sähköpyörätuoli, yskimisapu- ja hengitystukilaitteet, on tärkeä merkitys. Parantumattoman taudin eteneminen merkitsee käytännössä avun tarpeen ja hoitokustannusten voimakasta lisääntymistä (1).
Lääkkeitä, geeniterapiaa ja solunsiirtoja lihastauteihin kehitetään parhaillaan aktiivisesti, ja paljon kliinisiä lääketutkimuksia on meneillään. Lääkevalvontaviranomaiset eivät kohdista harvinaistautien lääkkeisiin samoja vaatimuksia kuin muihin lääkkeisiin, ja tämä on tiedostettu lääketeollisuudessa sillä seurauksella, että erittäin harvinaisiin sairauksiinkin kehitellään lääkkeitä. Soveltuvista lääkkeistä odotetaan saatavan huomattavia voittoja laittamalla niille tähtitieteellinen hinta.
Kun on punnittavana kalliin uuden hoidon käyttöönotto, herää kysymys, kuinka paljon kyseistä sairautta sairastavia on ja minkälaisen taakan tauti aiheuttaa. Sairauden aiheuttamalla taakalla voidaan tarkoittaa ennenaikaista kuolleisuutta, toimintakyvyn heikkenemisen astetta ja kestoa, kertyviä kustannuksia ja henkistä kärsimystä.
Kehittyneissä länsimaissa yhteiskunnan kannalta suurin taakka aiheutuu Duchennen lihasdystrofiasta, spinaalisesta lihasatrofiasta, ALS-taudista ja kroonisesta inflammatorisesta demyelinisoivasta polyneuropatiasta (CIDP) (2). Mutta onko tilanne sama Suomessa? Voidaanko ulkomaisista tutkimuksista tehdä päätelmiä vai eroavatko suomalaisten sairaudet muista merkittävästi? Miten saisimme selville tarkemmin lihastautipotilaiden määrän ja hoidon tarpeen?
Aineisto ja menetelmät
Selvityksessä käytettiin kirjallisuushakua PubMed- (Medline) ja EMBASE-tietokannoista ja hakusanoja "prevalence", "incidence" ja "epidemiology" yhdistettynä "neuromuscular disease" -hakusanaan tai yksittäisten tautien nimiin. Harvinaisten tautien luettelo, jonka kansainvälinen asiantuntijaverkosto Orphanet on laatinut, oli kolmantena tietolähteenä (3).
Taudin esiintyvyys- ja ilmaantuvuustietoja kerättiin priorisoiden tuoreimpia tuloksia, systemaattisten tautikohtaisten katsausten ja meta-analyysien tuloksia sekä Suomen tai Pohjoismaiden, Euroopan ja Yhdysvaltain tietoja.
Jos sairauden epidemiologiasta oli meta-analyysitietoa saatavilla, siinä ilmaistuja 95 %:n luottamusvälin arvoja käytettiin, kun arvioitiin tapausten pienintä ja suurinta määrää Suomessa. Jos meta-analyysiä ei ollut, otettiin huomioon pienin ja suurin Orphanetissa tai hyvissä julkaisuissa todettu esiintyvyys tai ilmaantuvuus. Tapausten määrän arvioimiseksi saatu luku kerrottiin pääsääntöisesti Suomen väkiluvulla eli 5,5 miljoonalla. Esiintyvyyden perusteella arvioidut potilaiden enimmäismäärät pyöristettiin lähimpään kymmeneen. X-kromosomaalisissa taudeissa huomioitiin sukupuoli.
Alle 16-vuotiaiden määränä pidettiin 1 miljoonaa. Ennen aikuisikää kuolemaan johtavien tautien kohdalla käytettiin ilmaantuvuutta tai syntymäprevalenssia, ja Suomessa vuosittain syntyvien lasten määränä pidettiin 50 000:ta, josta puolet poikia.
Kaikista lihastaudeista ei ollut riittävän tarkkaa tietoa saatavissa. Tautien yleisyysjärjestys laadittiin sen mukaan, miten paljon enimmillään kyseistä tautia sairastavia olisi julkaisujen tietojen perusteella laskettuna Suomessa.
Koska neuromuskulaaritaudeilla on usein monta eri nimeä ja kansainvälinen tautiluokitus ICD-10 on niiden osalta epätarkka, yhdistettiin taulukoissa selvyyden vuoksi taudin nimeen harvinaistautien luokittelun koodinumero, ORPHA-koodi, ja geneettisissä taudeissa myös perinnöllisten tautien luettelon MIM-koodi, jotta tilastojen kansainvälinen vertailu olisi helpompaa.
Tulokset
Kirjallisuuskatsauksen perusteella esiintyvyydeltään yleisimmät neuromuskulaaritaudit anatomisesti jaoteltuina ovat
1) selkäytimen etusarven motoneuronitaudeista amyotrofinen lateraaliskleroosi (ALS) (4,5,6,7), myöhään alkava spinaalinen motoneuronisairaus (LOSMoN) (8) ja spinaalinen ja bulbaarinen motoneuronitauti (SBMA) eli Kennedyn tauti (9,10)
2) perifeerisistä neuropatioista, jotka vaurioittavat sekä motorisia että sensoria hermoja, Charcot–Marie–Toothin taudin tyypit 1 ja 2 (CMT1 ja CMT2) (11,12,13), haurashermo-oireyhtymä (HNPP) (7,11) ja krooninen inflammatorinen polyneuropatia (CIDP) (14)
3) hermo-lihasliitoksen sairauksista myasthenia gravis (MG) (15)
4) lihaskudoksen sairauksista Suomessa myotonisen dystrofian tyypit 1 ja 2 (DM1 ja DM2) (16,17), Beckerin lihasdystrofia (BMD) (7,18), distaaliset myopatiat, etenkin tibiaalinen lihasdystrofia ja Welanderin distaalinen myopatia (19,20,21); yleisyysjärjestyksessä seuraavina ovat kasvo-hartiaseudun dystrofia (FSHD) (23), hartia-lantiodystrofiat (LGMD) (22,23), dermatomyosiitti (24), polymyosiitti (24,25), synnynnäinen myotonia (26,27) ja mitokondriotaudit (28).
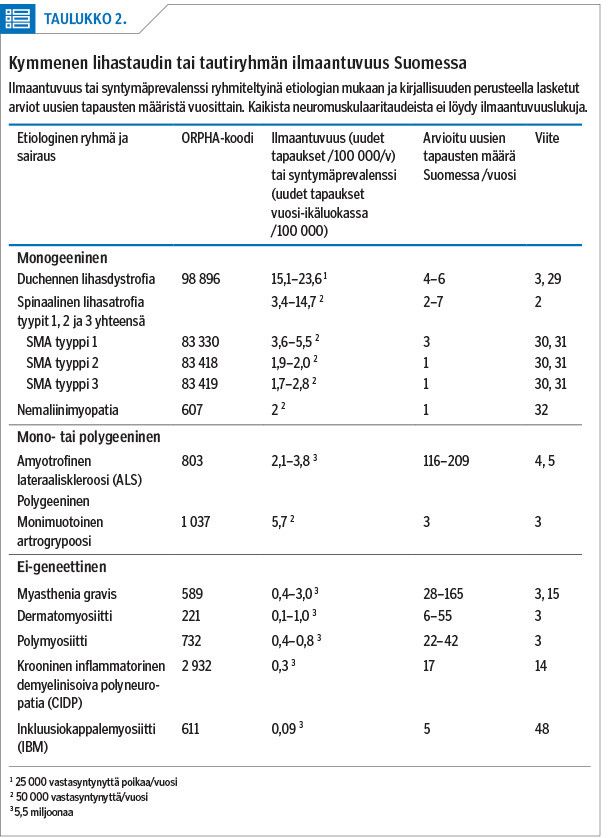
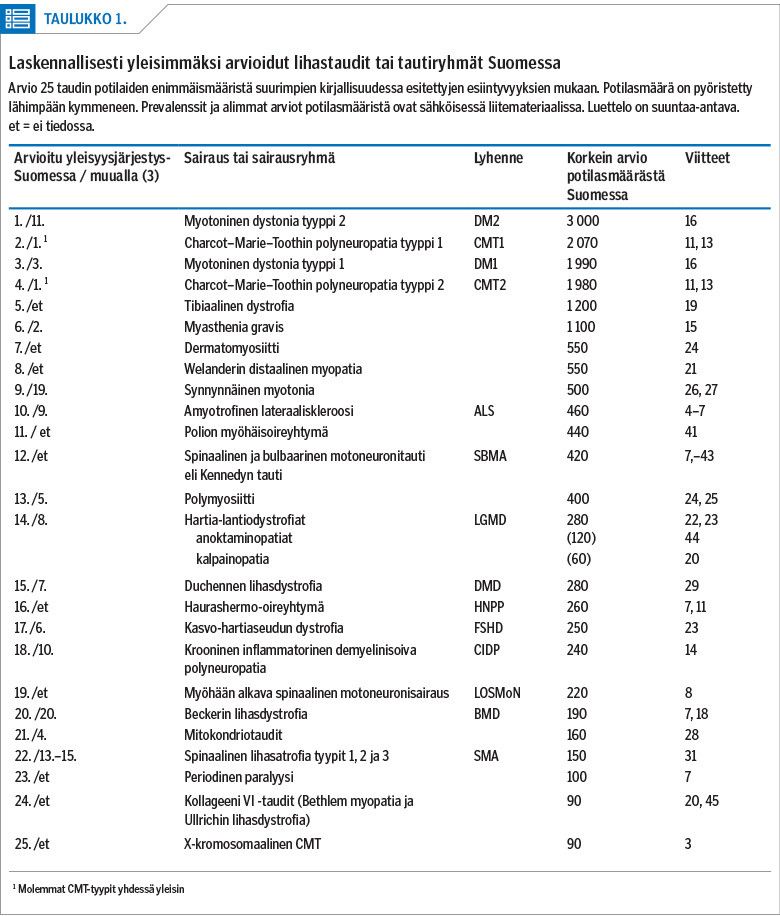
Julkaistujen esiintyvyyslukujen perusteella lasketut 25 yleisimmän lihassairauden tai lihassairausryhmän potilaiden enimmäismäärät on esitetty taulukossa 1. Kyseessä on puhtaasti laskennallinen arvio, eikä se perustu mihinkään sairaala-aineistoon. Yksityiskohtaisesti käytetyt esiintyvyydet sekä potilasmäärän pienin ja suurin arvio näissä ja eräissä vielä harvinaisemmissa taudeissa on esitetty liitetaulukossa artikkelin sähköisessä versiossa (Liitetaulukko 1, www.laakarilehti.fi > Sisällysluettelot > 10/2021).
Noiden 25 yleisimmän lihassairauden tai sairausryhmän joukkoon kuuluu monta sellaista, jonka esiintyvyys vaikuttaa olevan Suomessa suurempi kuin muissa maissa verrattaessa Orphanetin julkaisemiin lukuihin (taulukko 1). Suomessa arvioidaan esiintyvän suhteellisesti enemmän myotonisen dystrofian tyyppiä 2 (DM2), distaalisia myopatioita, LOSMoN-tautia, Kennedyn tautia ja synnynnäistä myotoniaa.
Neuromuskulaaritauteja sairastavien kokonaismäärän laskeminen on julkaistujen esiintyvyyslukujen suuren hajonnan vuoksi haastavaa. Yhteenlaskettu arvio potilasmäärästä Suomessa on pienimmillään 7 000 ja enimmillään 16 820 (taulukko 1).
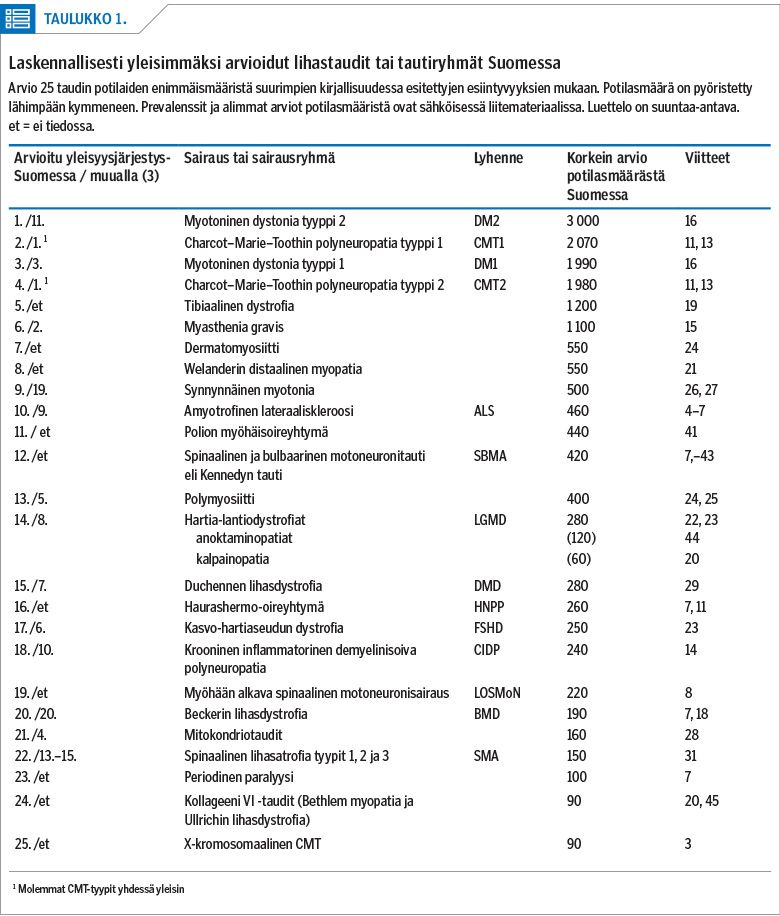
Uusien tapausten ilmaantuvuus ei ole suoraan verrannollinen esiintyvyyteen (taulukko 2). Aikuisiällä ilmenee eniten amyotrofista lateraaliskleroosia. Se voi etiologialtaan olla mono- tai polygeeninen. ALS:n ilmaantuvuus on suuri esiintyvyyteen nähden, koska se on vakava sairaus, jossa keskimääräinen elinaika diagnoosin jälkeen on vain 24–50 kuukautta (4,5). Aikuisiässä tehdään myös lisääntyvässä määrin uusia myotonisen dystrofian diagnooseja, kun geenitestaus yleistyy ja oireettomia tai vähäoireisiakin diagnosoidaan (16,17). Muista kuin geneettisistä taudeista yleisimmin diagnosoidaan myasthenia gravista (3,15), dermatomyosiittia (3), polymyosiittia (3) ja kroonista inflammatorista polyneuropatiaa (14) sekä inkluusiokappalemyosiittia (25), jotka ovat harvoin lasten sairauksia.
Charcot–Marie–Toothin taudin diagnooseja tehdään sekä lastenneurologian että aikuisneurologian klinikoissa. Oireiston vaikeusaste vaihtelee lievästä vaikeaan taudin eri alatyypeissä. Etiologialtaan monogeenisiä sairauksia, joita diagnosoidaan eniten lapsuudessa, ovat Duchennen lihasdystrofia (3,29), spinaalisen lihasatrofian eri alatyypit (2,30,31), nemaliinimyopatiat (32) sekä dystroglykanopatioihin lukeutuvat Walker–Warburgin oireyhtymä (3), FKRP-myopatia (fukutin-related protein) (3) ja lihas-silmä-aivo-oireyhtymä (MEB) (3). Vastasyntyneisyysaikana ilmenee myös etiologialtaan avoin, lähinnä polygeeninen monimuotoinen artrogrypoosi (3).
Pohdinta
Tautien esiintyvyydessä on tunnetusti maantieteellisiä eroja. Potilasmäärän arvioiminen muiden maiden lukujen tai monikansallisten aineistojen pohjalta voi johtaa harhaan. Suomen asutushistoriaan perustuva väestön geneettinen erityislaatuisuus näkyy siinä, että meillä tietyt taudit ovat yleisempiä kuin muualla.
Suomessa on voimakas perustajamutaation vaikutus useassa taudissa, mm. Charcot–Marie–Toothin taudin tyypissä 2(11), synnynnäisessä myotoniassa (26) ja tibiaalisessa dystrofiassa (19). Esimerkiksi synnynnäisen myotonian poikkeuksellisen korkean esiintyvyyden Pohjois-Suomessa selittää CLCN1-geenin variantti p.Arg894Ter, joka gnomAD-tietokannan mukaan on Suomessa viisi kertaa niin yleinen kuin muualla Euroopassa. Kennedyn taudissa perustajamutaatio lienee skandinaavista alkuperää (10). Toisaalta Suomessa on eräitä sairauksia hyvin vähän tai ei lainkaan, kuten mm. Pompen tautia tai Miyoshin myopatiaa (19).
Käytännön näkemys lihastautipotilaiden määrästä poikkeaa laskennallisista arvoista, koska etiologia on syystä tai toisesta avoin melko suurella osalla potilaista. Diagnostiikan parantaminen on selvästi tarpeen. Harvinaissairauksissa tavoitteeksi on asetettu, että diagnoosiin päästään yhden vuoden kuluessa (33,34). Velton vastasyntyneen diagnostiikassa sekin on liian pitkä aika, koska mitä varhaisemmin spinaalisen lihasatrofian tyypin 1 lääkehoito aloitetaan, sitä parempi on lopputulos (35). Lastenneurologien ja neurologien erikoistumiskoulutukseen tulisi kuulua riittävästi perehtymistä lihastauteihin.
Jos diagnoosi on epävarma, sitä tulisi pyrkiä tarkentamaan vastaanottokäynneillä. Täydentävistä tutkimuksista tärkeimpiä ovat genetiikan uudet menetelmät, etenkin geenipaneelit ja eksomisekvensointi. Asiantuntijakonsultaatioita ja verkostoja tarvitaan kotimaassa ja kansainvälisesti. Digitaaliset keinot konsultointiin ovat koko ajan parantuneet mm. Euroopan osaamiskeskusverkoston ERN:n ansiosta (https://ern-euro-nmd.eu/), mutta tiukka tietosuojalainsäädäntö asettaa toisaalta rajoituksia.
Antaako genomitieto oikeamman kuvan harvinaisten lihassairauksien esiintyvyydestä (36)? Jos kyseessä on yhden geenin tauti ja taudin periytymismalli ja tietyn variantin (mutaation) patogeenisuus tunnetaan, sen alleelitiheydestä voi laskea taudin esiintyvyyden väestössä. Laskelmia on tehty mm. hartia-lantiodystrofioiden esiintyvyydestä (37).
Perimästä (genotyypistä) ei kuitenkaan aina voi päätellä ilmiasua (fenotyyppiä), tässä tapauksessa sairautta, eikä päinvastoin. Taudin ilmenemisyleisyys (penetranssi) voi olla pienentynyt. Esimerkiksi myotonisen dystrofian oireet voivat olla hyvin lievät tai tibiaalisen dystrofian oireet manifestoitua niin korkeassa iässä, että niiden erottaminen normaaliin vanhenemiseen kuuluvasta lihasheikkoudesta on vaikeaa. Osassa mitokondriotaudeista mutaatio ei aiheuta lihasoireita (38). Sairauden takana voi olla useita geenejä tai ympäristötekijöiden ja geenien yhteisvaikutus. Tietyn geenin alleelin esiintyvyys on eri asia kuin kliinisiin oireisiin perustuva taudin esiintyvyys. Tarvitsemme jatkossakin kliinistä diagnoosia päätöksenteon tueksi.
Tämän katsauksen virhelähteistä yksi on se, ettei kyse ole systemaattisesta katsauksesta, vaan valikoidusta kirjallisuudesta, joka valaisee kysymystä lihastautien esiintyvyydestä Suomessa. Tutkimusten heterogeenisyys voi vääristää arvioita suuntaan tai toiseen ja näkyy potilasmäärän arvion suurena vaihteluvälinä. Toiseksi maantieteelliset tautien esiintymisen erot ja taudin esiintyminen tietyssä ikäryhmässä tai muussa rajatussa väestönosassa voivat vääristää tulosta. Kun sairaudet esiintyvät ryvästyminä tietyillä alueilla Suomessa, alueen tutkimuksen tuottamat esiintyvyysluvut johtavat yliarvioon, jos ne yleistetään koskemaan koko Suomea.
Jos sairaus lyhentää elinikää, tulee laskennallisesta potilasmäärästä todellisuuteen nähden yliarvio. Lisäksi myosiittien luokitteluun ei tarkkoja kriteereitä ole, diagnooseissa on päällekkäisyyttä ja inkluusiokappalemyosiittia arvellaan olevan piilossa (so. diagnosoimatta) senioriväestössä. Myosiittimaisia oireita esiintyy usein kriittisesti sairailla, mutta tapausten määrää on vaikea arvioida (39) eikä niitä ole tässä arviossa otettu mukaan. Statiinien käyttöön liittyvien tunnettujen, lihastaudin tapaisten haittavaikutusoireiden osuuttakaan ei ole laskettu arvioissa mukaan (40). Polion myöhäisoireyhtymä on maailmanlaajuisesti yleinen motoneuronisairaus (2), mutta meillä häviämässä. Vuonna 2019 Suomessa oli 436 polion jälkitilan vuoksi eläkkeensaajan vammaistukea saavaa (41), ja määrä pienenee tasaisesti vuosi vuodelta.
Merkittävin syy esiintyvyysarvion epätarkkuuteen ovat potilaat, joiden diagnoosina on G72.9 eli elektroneuromyografiaan (ENMG), lihasten magneettikuvaukseen tai lihasbiopsiaan perustuva näyttö lihastaudista, mutta tutkimukset ovat kesken.
Luotettavimman tiedon lihastautipotilaiden määrästä saisi poimimalla heistä tietyt perustiedot vastaanoton yhteydessä Kanta-arkistoon käyntitietoja tallennettaessa toisiolain (Laki sosiaali- ja terveystietojen toissijaisesta käytöstä 1491/2019) perusteella, vastaavaan tapaan kuin noin 12 000 MS-potilaan neurorekisteriin (www.neurorekisteri.fi). Tiedoista muodostettaisiin lihastautirekisteri, joka olisi sekä laaturekisteri että tutkimusrekisteri, kattaisi suunnilleen kaikki potilaat ja toimisi sairaanhoitopiirin tasolla. Valtakunnallisella tasolla koottaisiin eri sairaanhoitopiirien rekistereistä kansallinen lihastautirekisteri, joka toimisi viranomaisten valvonnassa.
Kansallisen lihastautirekisterin tehtävä olisi palvella tutkijoita, kliinikkoja, päätöksentekijöitä ja tietysti potilaita. Sen avulla lihastautipotilaiden hoidon laatua voitaisiin kehittää ja arvioida säännöllisesti. Kalliit uudet hoidot ovat jo alkaneet ja vaativat systemaattista seurantaa. Ainakin Duchennen lihasdystrofian, spinaalisen lihasatrofian ja ALS:n rekistereitä on kehitteillä eräissä sairaanhoitopiireissä. Asetusta terveydenhuollon valtakunnallisista laaturekistereistä valmistellaan parhaillaan sosiaali- ja terveysministeriössä.
Lopuksi
Lihastaudit yleistyvät, kun väestö vanhenee, koska monet niistä ilmenevät korkeassa iässä (1,42). Toisaalta vaikeasti sairaita lapsia, joiden hoito on kallista ja pitkäaikaista, pysyy tulevaisuudessa enemmän ja pitempään hengissä kuin nyt. Tietääksemme, kuinka paljon nämä trendit lisäävät yhteiskunnallista, taloudellista ja inhimillistä rasitusta, tarvitsemme tarkempaa tietoa potilasmääristä.
Lihastautia sairastavien määrän arvioiminen kirjallisuuden perusteella on haastavaa. Paras tapa saada tietoa todellisista esiintymisluvuista olisi lihastautipotilaiden kansallinen potilasrekisteri. Se olisi sekä tutkimus- että laaturekisteri ja auttaisi luomaan edellytyksiä korkeatasoiselle hoidolle ja kansainväliselle yhteistyölle. Tarkalla tiedolla lihastautien esiintyvyydestä Suomessa olisi merkitystä laadittaessa hoitosuosituksia ja pohdittaessa uusien, usein kalliiden hoitomuotojen käyttöönottoa. Kalliit hoidot ja niiden vaikuttavuus vaativat huolellisen seurannan. Vaikka yksittäiset neuromuskulaaritaudit ovat harvinaisia, kokonaisuutena ne ovat merkittävä harvinaistautien ryhmä Suomessa.
Jaana Lähdetie: Asiantuntijapalkkio (Lihastautiliitto ry). Matka- ja kokouskulut (Biogen, PTC Pharma, TREAT-NMD), TREAT-NMD-verkoston rekisterien Data Oversight Committeen Suomen edustaja ja Suomen TREAT-NMD Lihastautirekisterin vastuuhenkilö 2008–2018.
- 1
- Ryder S, Leadley RM, Armstrong A ym. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis 2017;12:79. doi: 10.1186/s13023-017-0631-3
- 2
- Bhatt JM. The epidemiology of neuromuscular disease. Neurol Clin 2016;34:999–1021.
- 3
- Orphanet Report Series, Rare Diseases Collection 2020;1:1–78. www.orpha.net
- 4
- Longinetti E, Regodon Wallin A, Samuelsson K ym. The Swedish motor neuron disease quality registry. Amyotroph Lateral Scler Frontotemporal Degener 2018;19:528–37.
- 5
- Longinetti E, Fang F. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol 2019;32:771–6.
- 6
- Benjaminsen E, Alstadhaug KB, Gulsvik M ym. Amyotrophic lateral sclerosis in Nordland County, Norway, 2000–2015: prevalence, incidence, and clinical features. Amyotroph Lateral Scler Frontotemporal Degener 2018;19:522–7.
- 7
- Lefter S, Hardiman O, Ryan AM. A population-based epidemiologic study of adult neuromuscular disease in the Republic of Ireland. Neurology 2017;88:304–13.
- 8
- Penttilä S, Jokela M, Bouquin H, Saukkonen AM, Toivanen J, Udd B. Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann Neurol 2015;77:163–72.
- 9
- Udd B, Juvonen V, Häkämies L ym. High prevalence of Kennedy’s disease in Western Finland – is the syndrome underdiagnosed? Acta Neurol Scand 1998;98:128–33.
- 10
- Lund A, Udd B, Juvonen V, Andersen PM ym. Multiple founder effects in spinal and bulbar muscular atrophy (SBMA, Kennedy disease) around the world. Eur J Human Genetics 2001;9:431–44.
- 11
- Marttila M, Kytövuori L, Helisalmi S ym. Molecular epidemiology of Charcot-Marie-Tooth disease in Northern Ostrobothnia, Finland: A population-based study. Neuroepidemiol 2017;49:34–9.
- 12
- Barreto LC, Oliveira FS, Nunes PS ym. Epidemiologic study of Charcot-Marie-Tooth Disease: A systematic review. Neuroepidemiol 2016;46:157–65.
- 13
- Braathen GJ, Sandb JC, Lobatob A, Høyerc H, Russella MB. Genetic epidemiology of Charcot–Marie–Tooth in the general population. Eur J Neurol 2011;18:39–48.
- 14
- Broers MC, Bunschoten C, Nieboer D, Lingsma HF. Incidence and prevalence of chronic inflammatory demyelinating polyradiculoneuropathy: A systematic review and meta-analysis. Neuroepidemiol 2019;52:161–72.
- 15
- Hehir KM, Silvestri NJ. Generalized myasthenia gravis: classification, clinical presentation, natural history, and epidemiology. Neurol Clin 2018;36:253–60.
- 16
- Suominen T, Bachinski LL, Auvinen S ym. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet 2011;19:776–82.
- 17
- Lindberg C, Bjerkne F. Prevalence of myotonic dystrophy type 1 in adults in Western Sweden. Neuromuscul Disord 2017;27:159–62.
- 18
- Mah JK, Korngut L, Dykeman, J, Day L, Pringsheim T, Nathalie J. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 2014;24:482–91.
- 19
- Palmio J, Jokela M, Sandell S, Suominen T, Penttilä S, Udd B. Distaaliset myopatiat - laajeneva kirjo erilaisia tauteja myös Suomessa. Duodecim 2016;132:1635–44.
- 20
- Norwood FL, Harling C, Chinnery PF ym. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009;132:3175–86.
- 21
- Hackmann P, Sarparanta J, Lehtinen S ym. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol 2013;73:500–9.
- 22
- Theadom A, Rodrigues M, Roxburgh R ym. Prevalence of muscular dystrophies: a systematic literature review. Neuroepidemiol 2014;43:259–68.
- 23
- Taghizadeh E, Rezaee M, Barreto GE, Sahebkar A. Prevalence, pathological mechanisms, and genetic basis of limb-girdle muscular dystrophies: A review. J Cell Physiol 2019;234:7874–84.
- 24
- Yang SH, Chang C, Lian ZX. Polymyositis and dermatomyositis – challenges in diagnosis and management. J Translat Autoimm 2019;2:100018. doi: 10.1016/j.jtauto.2019.100018
- 25
- Meyer A, Meyer N, Schaeffer M, Gottenberg J-E, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: A systematic review. Rheumatol (Oxford) 2015;54:50–63.
- 26
- Baumann P, Myllylä VV, Leisti J. Myotonia congenita in Northern Finland: an epidemiological and genetic study. J Med Genet 1998;35:293–6.
- 27
- Sun C, Tranebjaerg L, Torbergsen T, Holmgren G, Van Ghelue M. Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia. Eur J Hum Genet 2001;9:903–9.
- 28
- Gorman GS, Schaefer AM, Ng Y ym. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol 2015;77:753–9.
- 29
- Crisafulli S, Sultana J, Fontana A ym. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J Rare Dis 2020;15:141–60. doi: 10.1186/s13023-020-01430-8
- 30
- Verhaart IEC, Robertson A, Wilson IJ ym. Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis 2017;12:124–39. doi: 10.1186/s13023-017-0671-8.
- 31
- Darin N, Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord 2000;10:1–9.
- 32
- Cassandrini D, Trovato R, Rubegni A ym. Congenital myopathies: clinical phenotypes and new diagnostic tools. Review. Ital J Pediatr 2017;43:101–16.
- 33
- Austin CP, Cutillo CM, Lau LPL ym. Future of rare disease research 2017-2027: An IRDiRC perspective. Clin Transl Sci 2018;11:21–7. doi: 10.1111/cts.12500
- 34
- Harvinaisten sairauksien kansallinen ohjelma 2019-2023. Sosiaali- ja terveysministeriö. Raportteja ja muistioita 2019:49.
- 35
- Seppä-Moilanen M, Isohanni P, Lönnqvist T. Veltto imeväinen. Duodecim 2019;135:359–66.
- 36
- Valtioneuvosto. Parempaa terveyttä genomitiedon avulla. Kansallinen genomistrategia 2015. http://julkaisut.valtioneuvosto.fi/handle/10024/74514
- 37
- Liu W, Pajusalu S, Lake NJ ym. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet Med 2019;21:2312–20.
- 38
- Ahmed ST, Craven L, Russell OM, Turnbull DM, Vincent AE. Diagnosis and treatment of mitochondrial myopathies. Neurotherapeutics 2018;15:943–53.
- 39
- Friedrich O, Reid MB, Van den Berghe G ym. The sick and the weak: Neuropathies/Myopathies in the critically ill. Physiol Rev 2015;95:1025–109.
- 40
- Pohjola-Sintonen S, Julkunen H. Statiinien lihashaitat. Duodecim 2014;130:1622–7.
- 41
- Kallio-Laine K. Kelan tilastot. Henk.koht. tiedonanto 26.8.2020.
- 42
- Rose L, McKim D, Leasa D ym. Trends in incidence, prevalence, and mortality of neuromuscular disease in Ontario, Canada: A population-based retrospective cohort study (2003-2014). PLOS ONE March 26, 2019. doi: 10.1371/journal.pone.0210574
- 43
- Bertolin C, Querin G, Martinelli I, Pennuto M, Pegoraro E, Sorarù G. Insights into the genetic epidemiology of spinal and bulbar muscular atrophy: prevalence estimation and multiple founder haplotypes in the Veneto Italian region. Eur J Neurol 2019;26:519–24.
- 44
- Penttilä S, Palmio J, Suominen T ym. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology 2012;78:897–903.
- 45
- Collins D, Hicks A, Sarcozy A ym. Bethlem myopathy; new insights on prevalence, phenotypic variability and genetic heterogeneity. Neuromuscul Disord 2009;19:608.
- 46
- Parr JR, Andrew MJ, Finnis M, Beeson D, Vincent A, Jayawant S. How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch Dis Child 2014;99:539–42.
- 47
- Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004;14:635–49.
- 48
- Kaipiainen-Seppänen O, Aho K. Incidence of rare systemic rheumatic and connective tissue diseases in Finland. J Intern Med 1996;240:81–4.
Prevalence and incidence of neuromuscular diseases in Finland
Background Neuromuscular diseases are a heterogeneous group of rare diseases. It would be important to know the number of patients with neuromuscular diseases in Finland to be able to reduce the burden of disease for individuals and society. This review is based on recent research publications.
Methods A literature search on PubMed (Medline), EMBASE and Orphanet. Calculation of expected numbers (range) of patients based on prevalence and incidence data.
Results The number of neuromuscular disease patients in Finland with a population of 5.5 million is estimated to be at least 7000 and may be as high as 17000. The most prevalent diseases are likely to be myotonic dystrophy type 1 and type 2 and Charcot-Marie-Tooth disease type 1 and type 2. Amyotrophic lateral sclerosis, ALS, seems to have the highest incidence. Some diseases are more common in Finland than elsewhere in the world, due to the genetic history of Finnish people.
Conclusions Although rare, neuromuscular diseases are important disorders due to their severity. Without nationwide data it is impossible to know the exact number of affected persons and to plan optimal care. The new, expensive treatments that now are available require robust postmarketing evaluation and follow-up. A neuromuscular disease registry would be useful for researchers, clinicians and politicians and ultimately the patients themselves. It would help to improve diagnostics, treatment and research of these disorders both nationally and internationally.







{kind=link}
{kind=link}